OmicsHorizon 활용 사례
싱글셀, 멀티 오믹스, 통합 분석 등
오믹스호라이즌과 함께
원하는 분석을 수행하세요.
scRNA sequencing 분석,
Multi-omics 분석, 통합 분석 등
원하는 분석을 수행하세요.
scRNA sequencing 분석,
Multi-omics 분석, 통합 분석 등
원하는 분석을 수행하세요.
scRNA sequencing 분석
클러스터링부터 Marker gene 탐색, Cell type annotation까지 —
세포 수준의 이질성을 분석하며 인사이트를 쌓아가세요.
업로드한 논문의 Method 섹션을 기반으로
첨부한 Cell gene matrix 데이터를 분석해줘.
Results 섹션에 있는 Figure를 전부 포함해서
보여줘야 해.
Step 1 :
series_matrix.tsv 파일을 불러와 구조를 확인합니다.
Step 2 :
품질 관리(QC)를 수행하고 지표를 시각화합니다. (Violin plot)
Step 3 :
데이터를 정규화하고, 변동성이 높은 유전자를 선택한 뒤 시각화합니다. (Scatter plot)
Step 4 :
차원 축소(PCA, UMAP) 및 클러스터링을 수행한 후 시각화합니다. (UMAP plot)
Step 5 :
차등 발현 분석을 통해 클러스터 마커를 찾고 시각화합니다, (Dotplot/Matrixplot)
Step 6 :
궤적 추론(PAGA/DPT)을 수행하고 시각화합니다. (Trajectory plot)
1.
품질 관리 (Quality Control, QC)
가장 먼저 데이터의 전반적인 품질을 확인하고 분석에 적합하지 않은
저품질 세포와 유전자를 제거했습니다.
기준: 미토콘드리아 유전자 발현 비율(pct_counts_mt)이 5% 미만이고,
발현된 유전자 수(n_genes_by_counts)가 200개 이상 2500개 미만인 세포만 필터링했습니다.
결과: 필터링 후 983개의 세포와 11,780개의 유전자가 남았습니다.
(위 그림: 필터링 전후의 세포당 유전자 수, 총 발현량, 미토콘드리아 유전자 비율 분포를 보여주는 바이올린 플롯)

2.
정규화 및 특징 선택
(Normalization & Feature Selection)
세포 간의 시퀀싱 깊이 차이를 보정하고, 생물학적 의미가 큰 유전자를 선택했습니다.
방법
: 각 세포의 총 발현량을 10,000으로 맞춘 후 로그 변환(Log1p)을 수행했습니다.이후 세포 간 발현 변이가 큰 고변동 유전자(Highly Variable Genes, HVGs)를
추출했습니다.
결과
: 총 1,805개의 고변동 유전자가 식별되어 다운스트림 분석에 사용되었습니다.
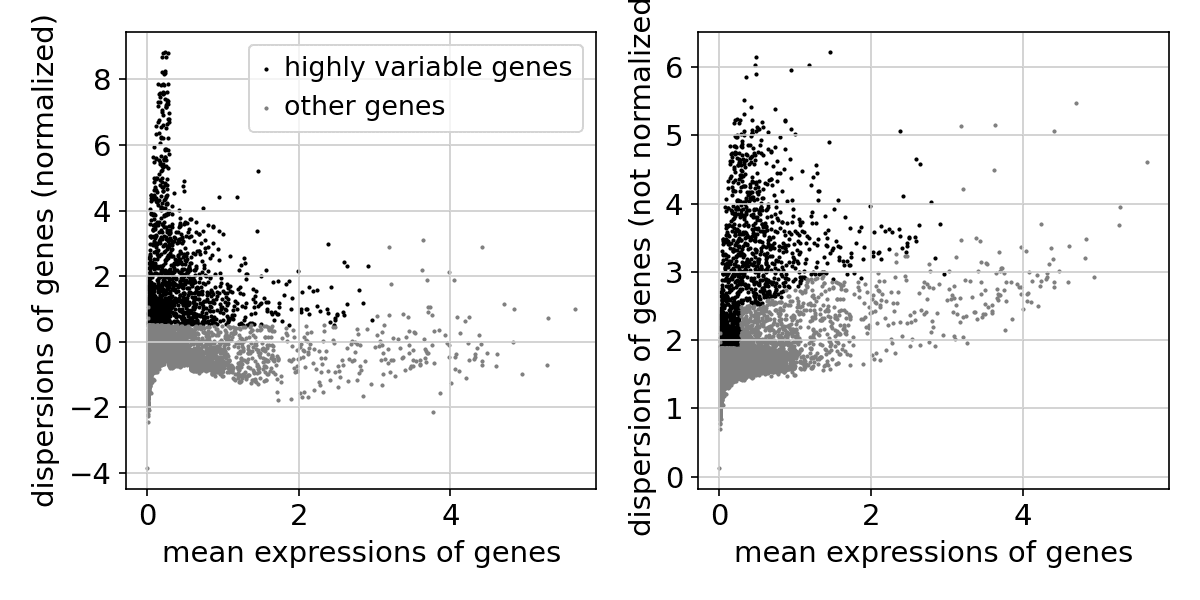
(위 그림: 유전자들의 평균 발현량 대비 분산을 보여주는 산점도. 검은색 점들이 선택된 고변동 유전자입니다.)
3.
차원 축소 및 군집화
(Dimensionality Reduction & Clustering)
복잡한 고차원 데이터를 2차원으로 시각화하고, 유사한 발현 패턴을 가진 세포들을
그룹화했습니다.
방법
: PCA를 통해 차원을 축소한 후, UMAP 알고리즘으로 2차원 공간에 투영했습니다.
세포 군집화는 Leiden 알고리즘(resolution=0.5)을 사용했습니다.
결과
: 총 5개의 뚜렷한 세포 군집(Cluster 0 ~ 4)이 식별되었습니다.
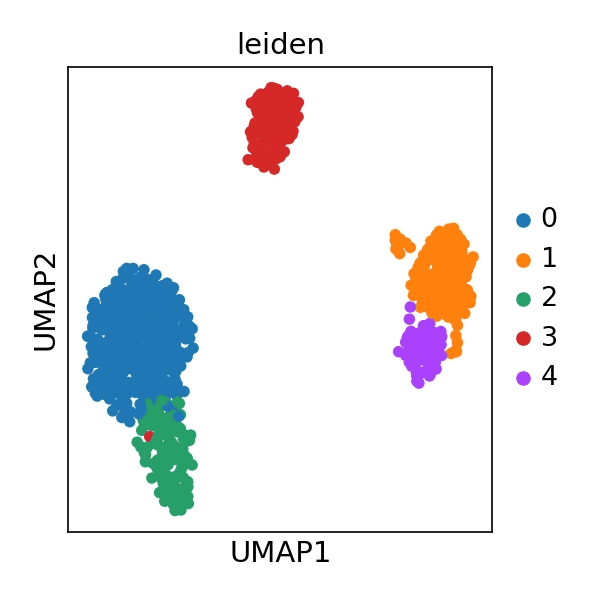
(위 그림: 5개의 군집이 각각 다른 색상으로 구분되어 2차원 공간에 매핑된 UMAP 플롯)
4.
차별 발현 분석 (Differential Expression Analysis)
각 군집의 생물학적 특성을 파악하기 위해 군집별 특이적 마커 유전자를 찾았습니다.
방법
: Wilcoxon rank-sum test를 사용하여 각 군집을 나머지 모든 군집과 비교했습니다.
결과
: 각 군집을 대표하는 상위 마커 유전자들이 식별되었습니다. (예: 군집 0의 경우 RPS12, LDHB, RPS25 등)

(위 그림: 각 군집별 상위 5개 마커 유전자의 평균 발현량(색상 진하기)과 발현 비율(점의 크기)을 보여주는 Dotplot)
scRNA sequencing 분석
클러스터링부터 Marker gene 탐색,
Cell type annotation까지 —
세포 수준의 이질성을 분석하며
인사이트를 쌓아가세요.
업로드한 논문의 Method 섹션을 기반으로 해서 첨부한
Cell gene matrix 데이터를
분석해줘.
Results 섹션에 있는 Figure를
전부 포함해서 보여줘야 해.
Step 1 :
series_matrix.tsv 파일을 불러와 구조를 확인합니다.
Step 2 :
품질 관리(QC)를 수행하고 지표를 시각화합니다.
(Violin plot)
(Violin plot)
Step 3 :
데이터를 정규화하고, 변동성이 높은 유전자를 선택한 뒤 시각화합니다.
(Scatter plot)
(Scatter plot)
Step 4 :
차원 축소(PCA, UMAP) 및 클러스터링을 수행한 후 시각화합니다.
(UMAP plot)
(UMAP plot)
Step 5 :
차등 발현 분석을 통해 클러스터 마커를 찾고 시각화합니다. (Dotplot/Matrixplot)
Step 6 :
궤적 추론(PAGA/DPT)을 수행하고 시각화합니다 (Trajectory plot).
1.
품질 관리
(Quality Control, QC)
가장 먼저 데이터의 전반적인 품질을 확인하고 분석에
적합하지 않은 저품질 세포와 유전자를 제거했습니다.
기준
: 미토콘드리아 유전자 발현 비율(pct_counts_mt)이 5% 미만이고,
발현된 유전자 수(n_genes_by_counts)가 200개 이상 2500개 미만인
세포만 필터링했습니다.결과
: 필터링 후 983개의 세포와 11,780개의 유전자가 남았습니다.
(위 그림: 필터링 전후의 세포당 유전자 수, 총 발현량, 미토콘드리아 유전자 비율 분포를 보여주는 바이올린 플롯)
2.
정규화 및 특징 선택
(Normalization & Feature Selection)
세포 간의 시퀀싱 깊이 차이를 보정하고, 생물학적 의미가 큰 유전자를 선택했습니다.
방법
: 각 세포의 총 발현량을 10,000으로 맞춘 후 로그 변환(Log1p)을
수행했습니다. 이후 세포 간 발현 변이가 큰 고변동 유전자(Highly Variable Genes, HVGs)를 추출했습니다.결과
: 총 1,805개의 고변동 유전자가 식별되어 다운스트림 분석에
사용되었습니다.
3.
차원 축소 및 군집화
(Dimensionality Reduction & Clustering)
복잡한 고차원 데이터를 2차원으로 시각화하고, 유사한 발현 패턴을 가진 세포들을
그룹화했습니다.
방법
: PCA를 통해 차원을 축소한 후, UMAP 알고리즘으로
2차원 공간에
투영했습니다.
세포 군집화는 Leiden 알고리즘(resolution=0.5)을사용했습니다.
결과
: 총 5개의 뚜렷한 세포 군집(Cluster 0 ~ 4)이 식별되었습니다.
(위 그림: 5개의 군집이 각각 다른 색상으로 구분되어 2차원 공간에 매핑된 UMAP 플롯)
4.
차별 발현 분석 (Differential Expression Analysis)
각 군집의 생물학적 특성을 파악하기 위해 군집별 특이적 마커 유전자를 찾았습니다.
방법
: Wilcoxon rank-sum test를 사용하여 각 군집을 나머지 모든 군집과 비교했습니다.
결과
: 각 군집을 대표하는 상위 마커 유전자들이 식별되었습니다. (예: 군집 0의 경우 RPS12, LDHB, RPS25 등)
(위 그림: 각 군집별 상위 5개 마커 유전자의 평균 발현량(색상 진하기)과 발현 비율(점의 크기)을 보여주는 Dotplot)
scRNA sequencing 분석
클러스터링부터 Marker gene 탐색, Cell type annotation까지 —
세포 수준의 이질성을 분석하며 인사이트를 쌓아가세요.
업로드한 논문의 Method 섹션을 기반으로
첨부한 Cell gene matrix 데이터를 분석해줘.
Results 섹션에 있는 Figure를 전부 포함해서
보여줘야 해.
Step 1 :
series_matrix.tsv 파일을 불러와 구조를 확인합니다.
Step 2 :
품질 관리(QC)를 수행하고 지표를 시각화합니다. (Violin plot)
Step 3 :
데이터를 정규화하고, 변동성이 높은 유전자를 선택한 뒤 시각화합니다.
(Scatter plot)
(Scatter plot)
Step 4 :
차원 축소(PCA, UMAP) 및 클러스터링을 수행한 후 시각화합니다. (UMAP plot)
Step 5 :
차등 발현 분석을 통해 클러스터 마커를 찾고 시각화합니다 .(Dotplot/Matrixplot)
Step 6 :
궤적 추론(PAGA/DPT)을 수행하고 시각화합니다. (Trajectory plot)
2.
정규화 및 특징 선택
(Normalization & Feature Selection)
세포 간의 시퀀싱 깊이 차이를 보정하고, 생물학적 의미가 큰
유전자를 선택했습니다.
방법:
각 세포의 총 발현량을 10,000으로 맞춘 후 로그 변환(Log1p)을 수행했습니다.
이후 세포 간 발현 변이가 큰 고변동 유전자(Highly Variable Genes, HVGs)를 추출했습니다.
결과:
총 1,805개의 고변동 유전자가 식별되어 다운스트림
분석에 사용되었습니다.
1.
품질 관리 (Quality Control, QC)
가장 먼저 데이터의 전반적인 품질을 확인하고 분석에 적합하지 않은 저품질
세포와 유전자를 제거했습니다.
기준:
미토콘드리아 유전자 발현 비율(pct_counts_mt)이
5% 미만이고, 발현된 유전자 수(n_genes_by_counts)가 200개
이상 2500개 미만인 세포만 필터링했습니다.
결과:
필터링 후 983개의 세포와 11,780개의 유전자가 남았습니다.
(위 그림: 필터링 전후의 세포당 유전자 수, 총 발현량, 미토콘드리아 유전자 비율 분포를 보여주는 바이올린 플롯)
3.
차원 축소 및 군집화
(Dimensionality Reduction & Clustering)
복잡한 고차원 데이터를 2차원으로 시각화하고, 유사한 발현 패턴을 가진 세포들을 그룹화했습니다.
방법: PCA를 통해 차원을 축소한 후, UMAP 알고리즘으로 2차원 공간에 투영했습니다. 세포 군집화는 Leiden 알고리즘(resolution=0.5)을 사용했습니다.
결과: 총 5개의 뚜렷한 세포 군집(Cluster 0 ~ 4)이 식별되었습니다.
(위 그림: 5개의 군집이 각각 다른 색상으로 구분되어 2차원 공간에 매핑된 UMAP 플롯)
4.
차별 발현 분석
(Differential Expression Analysis)
각 군집의 생물학적 특성을 파악하기 위해 군집별 특이적 마커 유전자를 찾았습니다.
방법
: Wilcoxon rank-sum test를 사용하여 각 군집을 나머지 모든
군집과 비교했습니다.
결과
: 각 군집을 대표하는 상위 마커 유전자들이 식별되었습니다.
(예: 군집 0의 경우 RPS12, LDHB, RPS25 등)
(위 그림: 각 군집별 상위 5개 마커 유전자의 평균 발현량(색상 진하기)과 발현 비율(점의 크기)을 보여주는 Dotplot)